Sindrome de Blau
| ||||

El Síndrome de Blau (BS). Es un término médico usado para describir una combinación de síntomas que afectan principalmente la piel, las articulaciones y los ojos aunque tiene un amplio espectro clínico cuyos síntomas pueden ser inespecíficos o tener afectación orgánica múltiple.
Sumario
Caracterización
El síndrome de Blau (BS) es una enfermedad inflamatoria sistémica poco frecuente que se caracteriza por la aparición temprana de artritis granulomatososa, uveítis y erupciones en la piel. En la actualidad, el BS se refiere tanto a la forma familiar como a la esporádica (anteriormente sarcoidosis de aparición temprana) de la misma enfermedad. El término propuesto de artritis granulomatosa pediátrica está actualmente en cuestión, ya que no representa la naturaleza sistémica de la enfermedad. Pueden verse afectados otros órganos y presentar fiebre intermitente.
Nombres alternativos
Granulomatosis familiar juvenil sistémica / Granulomatosis artrocutaneouveal / Jabs, Síndrome de Blau / Artritis inflamatoria granulomatosa, Dermatitis y uveitis familiar / Granulomatosis familiar tipo Blau.
Prevalencia
Aunque su prevalencia exacta es desconocida se calcula va de 10–40 casos por cada 100 mil personas. Se ha observado heterogenicidad entre grupos étnicos y raciales. Afecta a personas de los dos sexos, y de casi todas las edades, razas y sitios geográficos; las hembras son más susceptibles que los varones. Casi todos los casos presentan el mal entre los 20–40 años pero puede presentarse en niños incluso en lactantes.La máxima incidencia se encuentra en niños de 8 a15 años. Se han descrito cientos de casos de índole familiar.
Signos y síntomas
Los principales síntomas de la enfermedad son la tríada de artritis, dermatitis y uveítis. Los síntomas iniciales de la piel comprenden desde un exantema típico, con lesiones pequeñas y redondeadas de color variable, rosa pálido a bronceado, hasta un eritema intenso. Durante el transcurso de los años, la erupción aumenta y disminuye. La artritis es la manifestación más frecuente, iniciándose en la primera década de la vida. Al comienzo, se observa inflamación articular con movilidad limitada.
Con el tiempo, puede dar lugar a limitación del movimiento, deformidades y erosiones. La uveítis (inflamación del iris) es la manifestación más peligrosa, ya que suele estar asociada con complicaciones (cataratas, aumento de la presión intraocular) y puede producir descenso de la visión si no se trata. Además, la inflamación granulomatosa puede afectar a un amplio espectro de otros órganos, causando también otros síntomas como una menor funcionalidad de los pulmones o riñones, aumento te la tensión arterial o fiebre recurrente.
Las erupciones cutáneas aparecen a la edad de 1 mes en la cara y luego se extienden al tronco. Los pacientes pueden tener episodios intermitentes de lesiones cutáneas que se curan sin tratamiento. Las manifestaciones en las articulaciones generalmente comienzan antes de los 10 años con protuberancias indoloras como quistes en la parte posterior de los pies y las muñecas. Le sigue artritis simétrica de muñecas, tobillos y algunas veces codos. La incapacidad grave no suele producirse hasta la edad de 40-50 años. Una iridociclitis granulomatosa insidiosa y una uveítis posterior pueden convertirse en una panuveítis destructiva grave. Con el tiempo se pueden producir nódulos característicos en iris, sinequias focales, cataratas, aumento de la presión intraocular y precipitados caracteristicos en el limbo. Las afectaciones posteriores incluyen vitritis, coroiditis multifocal, vasculopatía retiniana y edema del nervio óptico. Se observa una pérdida visual significativa en el 20-30% de individuos afectados. El espectro de manifestaciones clínicas incluye fiebre, hipertensión sistémica y pulmonar maligna, vasculitis granulomatosa de grandes vasos e inflamación granulomatosa de hígado, riñones y pulmón.
Causas
El síndrome de Blau es una enfermedad genética. Está causado por mutaciones heredadas o de novo en el gen NOD2, gen responsable (sinónimo con CARD15), que codifica para una proteína con una función en la respuesta del sistema inmunitario y responsable de alteraciones en la respuesta inmune innata, la inflamación y la muerte celular. Si este gen es portador de una mutación, como ocurre en el Síndrome de Blau, la proteína no funciona correctamente y los pacientes experimentan inflamación crónica con formación de granulomas en varios tejidos y órganos del cuerpo. Los granulomas son grupos de células características asociadas con la inflamación que pueden alterar el normal funcionamiento de diversos tejidos y órganos. A partir de estudios de transfección, se ha propuesto que las mutaciones NOD2 causan la activación del factor nuclear kappa B que es a su vez un regulador de la transcripción de citoquinas proinflamatorias.
Se hereda como una enfermedad autosómica dominante (lo que significa que no está ligada al sexo y que al menos un progenitor debe mostrar síntomas de la enfermedad). Este tipo de transmisión significa que para tener el Síndrome de Blau, una persona necesita solamente un gen mutado, ya sea del padre o de la madre. En la SIP, la forma esporádica de la enfermedad, la mutación surge en el paciente por sí mismo, y los dos progenitores están sanos. Si un paciente es portador del gen, sufrirá la enfermedad. Si un paciente tiene el Síndrome de Blau, existe un 50 % de posibilidades de que su hijo o hija la sufra.
Posibles complicaciones
El BS es una enfermedad crónica y progresiva con un espectro de gravedad variable y a menudo impredecible. En los casos con manifestaciones amplias, la esperanza de vida puede ser reducida. La uveítis tiene un pronóstico desfavorable.
Diagnóstico
El diagnóstico descansa en gran medida en la demostración de la inflamación granulomatosa no caseificante con células epiteliales y células gigantes multinucleadas en biopsia sinovial, conjuntival, o cutánea, y test genéticos para mutaciones en el gen NOD2.
El diagnóstico diferencial incluye poliartritis y artritis juvenil idiopática de origen sistémico (JIA), inflamación granulomatosa asociada con inmunodeficiencias primarias, y vasculitis granulomatosa sistémica. En pacientes con inflamación granulomatosa, deben excluirse infecciones crónicas especialmente con micobacterias y hongos.
El diagnóstico prenatal y los test genéticos prenatales se realizan en raras ocasiones.
Prevención
El niño tiene la enfermedad porque porta el gen que causa el Síndrome de Blau. En la actualidad, la enfermedad no puede prevenirse pero los síntomas pueden tratarse.
Tratamiento
- Se han utilizado con resultados positivos los esteroides: la prednisona a 1 mg/kg./día durante 6 semanas y luego a 0,5 mg/kg/día en días alternos por 4 semanas.
- Los esteroides inhalados (budesonida, fluticasona), pueden ser útiles en pacientes con afectación pulmonar y tos importante sin que tengan efectos sobre la enfermedad sistémica. Recientemente se ha reportado el uso de esteroides tópicos e intralesionales en las lesiones cutáneas.
- El metotrexato a 15 mg/m2, repartidos en 3 o 4 subdosis durante 2 días consecutivos a la semana es un tratamiento que ha mostrado efectividad en pacientes resistentes a lo esteroides.
- La ciclosporina supone una alternativa de tratamiento al uso del metotrexato como coadyuvante del tratamiento esteroideo.
- La introducción de anticuerpos monoclonales anti-TNF (infliximab y adalimumab) pueden constituir un importante avance terapéutico en el tratamiento del BS; sin embargo, el efecto sobre la actividad de la uveítis puede ser menos convincente.
Galería
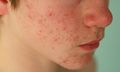
Cara
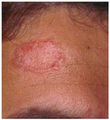
Frente

Lengua
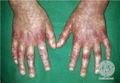
Manos
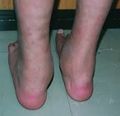
Piernas

Rodillas

Gluteos

Cuerpo, ojos, manos








Fuente
- Sarcoidosis. Disponible en:BVS Cuba. Consultado el 16 de marzo del 2017.
- Síndrome de Blau. Disponible en:Nomidalliance. Consultado el 16 de marzo del 2017.
- Sarcoidosis pediátrica. Disponible en:BVS Cuba. Consultado el 17 de marzo del 2017.
- Síndrome de Blau. Disponible en:ENFERMEDADES RARAS. Consultado el 17 de marzo del 2017.
- Síndrome de Blau. Disponible en:Reumaped. Consultado el 17 de marzo del 2017.
- Sarcoidosis. Disponible en:BVS. Consultado el 17 de marzo del 2017.